The reaction performed in this experiment was successful until the end of step 3. The end product of step 2, N-(4-methyl)-phenyloxyphthalimide, was produced precisely with a yield of 80%. Based off the H-NMRs taken after the completion of step 2, it was seen that N-(4-methyl)-phenyloxyphthalimide was definitely produced, however the compound consisted of several impurities. In both the H-NMR scans taken, there were several extra peaks proving that there were impurities in the compound.
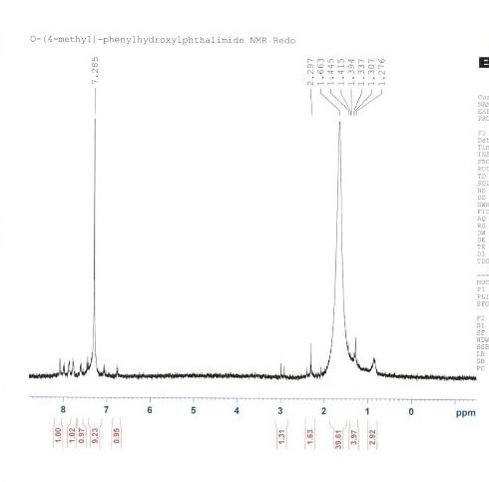
Figure 1 shows the H-NMR of the first attempt at synthesizing the product of step 2. This H-NMR proves that the compound produced was very impure. We can recognize a few of the peaks, however most of them are unidentifiable. For example, the very large peak that can be found between 7 and 8 parts per million can be identified as the chloroform used to take this H-NMR. The peaks to the left of the chloroform-d peak are supposed to correspond to the hydrogens on the compound, but the H-NMR of the compound taken had too many impurities. The synthesis of N-(4-methyl)-phenyloxyphthalimide contained several errors and deviations from the procedure given. For example, the product of step one, the diaryliodonium triflate, is unstable and shouldn’t be left unused for too long. When we finished step one, we allowed the compound to sit overnight in the refrigerator, instead of for half an hour, and continued onto step two the next day. This most likely caused the product we came back to the next day to be something that wasn’t diaryliodonium trifalte. Other errors include accidently spilling the compounds that were synthesized. This may have affected the scan taken of the compound in ways such as new reactions and different yields.
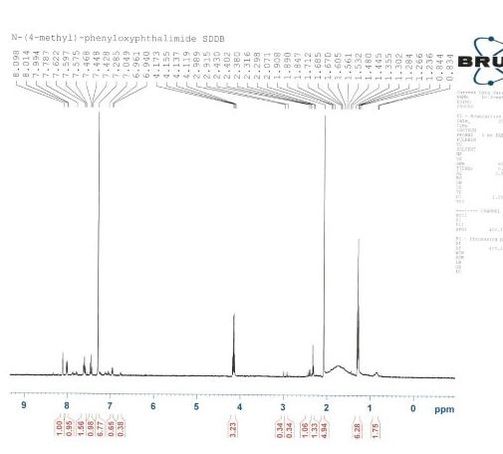
Figure 2 shows an H-NMR of the second attempt at synthesizing the compound after step 2. In the H-NMR (figure two), chloroform-d was directly added to the round bottom flask. This made the solution very concentrated with our compound. Since the solution was very concentrated, higher peaks were expected between 7 and 8 parts per million. However, since the peaks were small, it was concluded that the amount of N-(4-methyl)-phenyloxyphthalimide produced was very small. This also helped to explain that the yield of pure N-(4-methyl)-phenyloxyphthalimide produced was minuscule and most of the compound consisted of impure compounds. Several other peaks were also identifiable, mainly between 7.4 and 8.2 parts per million. These peaks corresponded with the hydrogen groups on the compound. The peak caused by the methyl group was also identified to be at 2.3. Although not much of N-(4-methyl)-phenyloxyphthalimide was produced, it was concluded that it was still synthesized successfully and can be synthesized at higher yields in a more professional lab setting.
Figure 1 shows the H-NMR of the first attempt at synthesizing the product of step 2. This H-NMR proves that the compound produced was very impure. We can recognize a few of the peaks, however most of them are unidentifiable. For example, the very large peak that can be found between 7 and 8 parts per million can be identified as the chloroform used to take this H-NMR. The peaks to the left of the chloroform-d peak are supposed to correspond to the hydrogens on the compound, but the H-NMR of the compound taken had too many impurities. The synthesis of N-(4-methyl)-phenyloxyphthalimide contained several errors and deviations from the procedure given. For example, the product of step one, the diaryliodonium triflate, is unstable and shouldn’t be left unused for too long. When we finished step one, we allowed the compound to sit overnight in the refrigerator, instead of for half an hour, and continued onto step two the next day. This most likely caused the product we came back to the next day to be something that wasn’t diaryliodonium trifalte. Other errors include accidently spilling the compounds that were synthesized. This may have affected the scan taken of the compound in ways such as new reactions and different yields.
Figure 2 shows an H-NMR of the second attempt at synthesizing the compound after step 2. In the H-NMR (figure two), chloroform-d was directly added to the round bottom flask. This made the solution very concentrated with our compound. Since the solution was very concentrated, higher peaks were expected between 7 and 8 parts per million. However, since the peaks were small, it was concluded that the amount of N-(4-methyl)-phenyloxyphthalimide produced was very small. This also helped to explain that the yield of pure N-(4-methyl)-phenyloxyphthalimide produced was minuscule and most of the compound consisted of impure compounds. Several other peaks were also identifiable, mainly between 7.4 and 8.2 parts per million. These peaks corresponded with the hydrogen groups on the compound. The peak caused by the methyl group was also identified to be at 2.3. Although not much of N-(4-methyl)-phenyloxyphthalimide was produced, it was concluded that it was still synthesized successfully and can be synthesized at higher yields in a more professional lab setting.
(Figure 1)
(Figure 2)
